![]()
![]()
using Markdown
using TensorKit
using MPSKit
using MPSKit: infinite_temperature_density_matrix
using MPSKitModels
using QuadGK: quadgk
using SpecialFunctions: ellipe
using Plots
using LinearAlgebra
using BenchmarkFreeFermionsFinite temperature XY model
This example shows how to simulate the finite temperature behavior of the XY model in 1D. Importantly, the Hamiltonian can be diagonalized in terms of fermionic creation and annihilation operators. As a result, many properties have analytical expressions that can be used to verify our results. Here, we use BenchmarkFreeFermions.jl to compare our results.
Here we will consider the anti-ferromagnetic (
Parameters
J = 1 / 2
T = ComplexF64
symmetry = U1Irrep
function XY_hamiltonian(
::Type{T} = ComplexF64, ::Type{S} = Trivial; J = 1 / 2, N
) where {T <: Number, S <: Sector}
spin = 1 // 2
term = J * (S_xx(T, S; spin) + S_yy(T, S; spin))
lattice = isfinite(N) ? FiniteChain(N) : InfiniteChain(1)
return @mpoham begin
sum(nearest_neighbours(lattice)) do (i, j)
return term{i, j}
end
end
endXY_hamiltonian (generic function with 3 methods)Diagonalization of the Hamiltonian
The Hamiltonian can be diagonalized through a Bogoliubov transformation, leading to the following expression for the ground state energy The Hamiltonian can be diagonalized in terms of fermionic creation and annihilation operators, leading to the following expression in terms of an incomplete elliptic integral of the second kind.
The derivation, via a Jordan-Wigner transformation to free fermions followed by a Bogoliubov rotation, can be found in Lieb, Schultz & Mattis, Ann. Phys. 16, 407 (1961).
function groundstate_energy(J, N)
isfinite(N) || return -J / π
T = diagm(1 => J / 2 * ones(N - 1), -1 => J / 2 * ones(N - 1))
ϵ = SingleParticleSpectrum(T)
return Energy(ϵ, Inf, 0) / N
endgroundstate_energy (generic function with 1 method)Exact diagonalization
We can check our results by comparing them to the exact diagonalization of the Hamiltonian.
N_exact = 6
H = open_boundary_conditions(XY_hamiltonian(T, symmetry; J, N = Inf), N_exact)
H_dense = convert(TensorMap, H);
vals = eigvals(H_dense)[one(symmetry)] ./ N_exact
groundstate_energy(J, N_exact)
println("Numerical:\t", minimum(real(vals)))
println("Exact (N=$(N_exact)):\t", groundstate_energy(J, N_exact))
println("Exact (N=Inf):\t", groundstate_energy(J, Inf))Numerical: -0.1455816336431223
Exact (N=6): -0.14558163364312227
Exact (N=Inf): -0.15915494309189535Finite MPS
If we wish to increase the system size, we can use the finite MPS representation.
N = 32
H = XY_hamiltonian(T, symmetry; J, N)
D = 64
V_init = symmetry === Trivial ? ℂ^32 : U1Space(i => 10 for i in -1:(1 // 2):1)
psi_init = FiniteMPS(N, physicalspace(H, 1), V_init)
trscheme = truncrank(D)
psi, envs, = find_groundstate(psi_init, H, DMRG2(; trscheme, maxiter = 5));
E_0 = expectation_value(psi, H, envs) / N
println("Numerical:\t", real(E_0))
println("Exact (N=$N):\t", groundstate_energy(J, N))
println("Exact (N=Inf):\t", groundstate_energy(J, Inf))[ Info: DMRG2 1: obj = -5.004084869795e+00 err = 9.8826020061e-01 time = 1.04 min
[ Info: DMRG2 2: obj = -5.004096937587e+00 err = 1.1541480959e-06 time = 0.42 sec
[ Info: DMRG2 3: obj = -5.004096975044e+00 err = 2.5015824967e-09 time = 0.72 sec
[ Info: DMRG2 conv 4: obj = -5.004096975044e+00 err = 1.8118839762e-13 time = 1.07 min
Numerical: -0.15637803047010948
Exact (N=32): -0.15637803047254015
Exact (N=Inf): -0.15915494309189535Finite temperature properties
To go beyond the ground state, we can extract several properties at finite temperature by computing the partition function. This is given by
where
Given the partition function, we can compute the free energy as
We can also compute observables using
In particular, we can compute the energy as
Finally, the specific heat can be computed as
Luckily, the partition function can be computed analytically for the XY model. The resulting expression is
This expression follows from the same free-fermion diagonalization as the ground-state energy above: each single-particle mode
function partition_function(β::Number, J::Number, N::Number)
T = diagm(1 => J / 2 * ones(N - 1), -1 => J / 2 * ones(N - 1))
ϵ = SingleParticleSpectrum(T)
return LogPartition(ϵ, β, 0) / N
end
function free_energy(β, J, N)
T = diagm(1 => J / 2 * ones(N - 1), -1 => J / 2 * ones(N - 1))
ϵ = SingleParticleSpectrum(T)
return FreeEnergy(ϵ, β, 0) / N
end
βs = 0.0:0.2:8.0
Z_analytic = partition_function.(βs, J, N);
F_analytic = free_energy.(βs, J, N);MPO approach
We can numerically compute the partition function by explicitly computing the trace of the time-evolution operator. To that end, we first need to build the time-evolution operator
In order to build the time-evolution operator, we can repurpose the make_time_mpo function, which constructs the time-evolution operator for the ground state. However, since we are interested in TaylorCluster algorithm.
expansion_orders = 1:3
function logpartition_taylor(β, H; expansion_order)
dτ = im * β
expH = make_time_mpo(H, dτ, TaylorCluster(; N = expansion_order))
return log(real(tr(expH))) / length(H)
end
Z_taylor = map(Iterators.product(βs, expansion_orders)) do (β, expansion_order)
@info "Computing β = $β at order $expansion_order"
return logpartition_taylor(β, H; expansion_order)
end
F_taylor = -(1 ./ βs) .* Z_taylor
p_taylor = let
labels = reshape(
map(expansion_orders) do N
return "Taylor N=$N"
end, 1, :
)
p1 = plot(
βs, Z_analytic; label = "analytic", title = "Partition function",
xlabel = "β", ylabel = "Z(β)"
)
plot!(p1, βs, Z_taylor; label = labels)
p2 = plot(
βs, F_analytic; label = "analytic", title = "Free energy",
xlabel = "β", ylabel = "F(β)"
)
plot!(p2, βs, F_taylor; label = labels)
plot(p1, p2)
end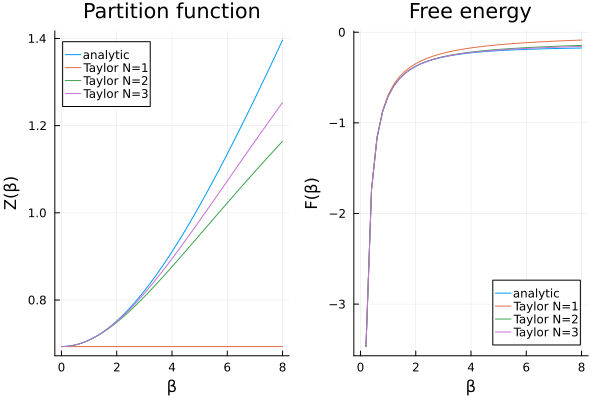
Some observations:
The first order approximation fails to capture the behavior of the partition function.
The higher order approximations are in good agreement with the analytical result, as long as
is not too large. The computational cost of the approximations does not depend on
, but on the order of the approximation.
To address the first point, we can have a look at the particular form of the time-evolution operator. Here we see that for this particular Hamiltonian, all the terms with factors
Therefore, we will exclude the first order approximation from now on. Zooming in on the differences with the analytical result, we find:
expansion_orders = 2:3
Z_taylor = Z_taylor[:, 2:end]
F_taylor = F_taylor[:, 2:end]
p_taylor_diff = let
labels = reshape(
map(expansion_orders) do N
return "Taylor N=$N"
end, 1, :
)
p1 = plot(
βs, abs.(Z_taylor .- Z_analytic);
label = labels, title = "Partition function error",
xlabel = "β", ylabel = "ΔZ(β)", legend = :topleft
)
p2 = plot(
βs, abs.(F_taylor .- F_analytic); label = labels,
xlabel = "β", ylabel = "ΔF(β)", title = "Free energy error", legend = :topleft
)
plot(p1, p2)
end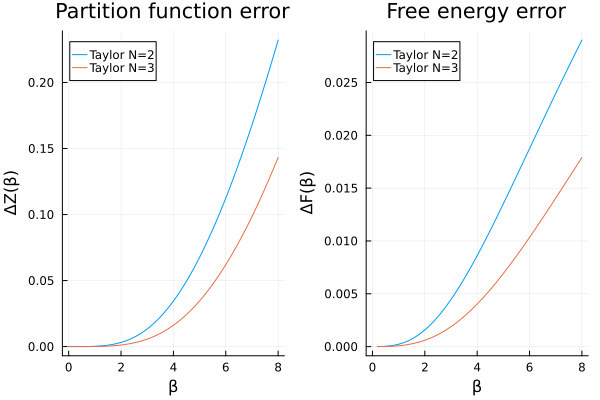
We can now clearly see that, somewhat unsurprisingly, the error increases the larger
However, there is a trick we can use to improve our results slightly. To that end, we first rewrite the partition function as
In other words, we can compute the partition function at
double_logpartition(ρ₁, ρ₂ = ρ₁) = log(real(dot(ρ₁, ρ₂))) / length(ρ₁)
function logpartition_taylor2(β, H; expansion_order)
dτ = im * β / 2
expH = make_time_mpo(H, dτ, TaylorCluster(; N = expansion_order))
return double_logpartition(expH)
end
Z_taylor2 = map(Iterators.product(βs, expansion_orders)) do (β, expansion_order)
@info "Computing β = $β at order $expansion_order"
return logpartition_taylor2(β, H; expansion_order)
end
F_taylor2 = -(1 ./ βs) .* Z_taylor2
p_taylor2_diff = let
labels = reshape(
map(expansion_orders[2:end]) do N
return "Taylor N=$N"
end, 1, :
)
p1 = plot(
βs, abs.(Z_taylor2 .- Z_analytic);
label = labels, title = "Partition function error",
xlabel = "β", ylabel = "ΔZ(β)", legend = :topleft
)
p2 = plot(
βs, abs.(F_taylor2 .- F_analytic); label = labels,
xlabel = "β", ylabel = "ΔF(β)", title = "Free energy error", legend = :topleft
)
plot(p1, p2)
end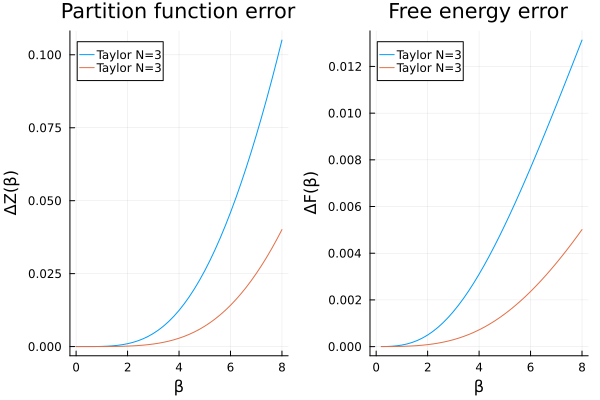
MPO multiplication approach (linear)
While the Taylor series approach is useful, we can only push that so far, since we are always expanding around
Multiplying two MPOs exactly would lead to an exponential growth in bond dimensions, but we can make use of standard MPS techniques to keep the bond dimensions under control. To achieve this, we can reinterpret the density matrix as an MPS with two physical indices. Then, we have some control over the approximations we make by tuning the maximal bond dimension.
Here, we swich to a logarithmic scale for the errors to better illustrate the results.
Warning
Using MPS techniques to approximate the multiplication of density matrices does not necessarily inherit all of the nice properties of approximating MPS. In particular, the truncation of the MPO is now happening in the Frobenius norm, rather than the operator norm. While for small truncations this might still work, this is not guaranteed to be the case for larger truncations. As a result, the truncated object might not be positive semidefinite, spoiling its interpretation as a density matrix.
Z_mpo_mul = zeros(length(βs))
D_max = 64
# first iteration: start from high order Taylor expansion
ρ₀ = make_time_mpo(H, im * βs[2] / 2, TaylorCluster(; N = 3))
Z_mpo_mul[1] = Z_taylor[1]
Z_mpo_mul[2] = double_logpartition(ρ₀)
# subsequent iterations: multiply by ρ₀
ρ_mps = convert(FiniteMPS, ρ₀)
for i in 3:length(βs)
global ρ_mps
@info "Computing β = $(βs[i])"
ρ_mps, = approximate(
ρ_mps, (ρ₀, ρ_mps), DMRG2(; trscheme = truncrank(D_max), maxiter = 10)
)
Z_mpo_mul[i] = double_logpartition(ρ_mps)
end
F_mpo_mul = -(1 ./ βs) .* Z_mpo_mul
p_mpo_mul_diff = let
labels = reshape(
map(expansion_orders) do N
return "Taylor N=$N"
end, 1, :
)
p1 = plot(
βs, abs.(Z_taylor2 .- Z_analytic);
label = labels, title = "Partition function error",
xlabel = "β", ylabel = "ΔZ(β)", legend = :bottomright, yscale = :log10
)
plot!(
p1, βs, abs.(Z_mpo_mul .- Z_analytic);
label = "MPO multiplication"
)
p2 = plot(
βs, abs.(F_taylor2 .- F_analytic); label = labels,
xlabel = "β", ylabel = "ΔF(β)", title = "Free energy error", legend = nothing,
yscale = :log10
)
plot!(
p2, βs, abs.(F_mpo_mul .- F_analytic);
label = "MPO multiplication"
)
plot(p1, p2)
end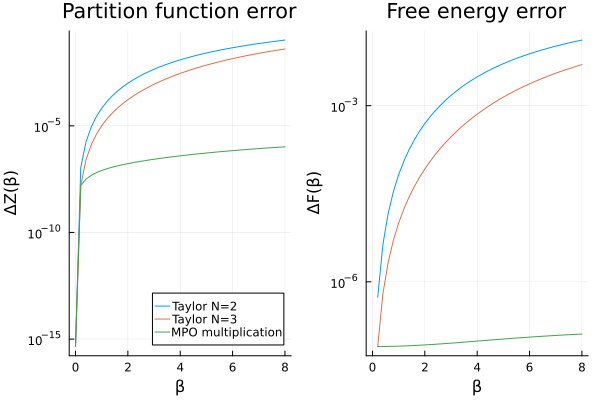
This approach clearly improves the accuracy of the results, indicating that we can indeed compute partition functions at larger
We also have to be careful with the accuracy of our results. In particular, the error in the partition function will accumulate over the iterations, which might turn the results into garbage. Typically, the entanglement entropy of the density matrix is a good measure of the required bond dimension, and we can use this to tune the maximal bond dimension.
Apart from the bond dimension, we have two other parameters to tune: the accuracy of the initial density matrix, and the size of the step. The accuracy of the initial density matrix can be improved by increasing the order of the Taylor expansion, but this will result in a larger MPO bond dimension. On the other hand, if we improve the accuracy of the initial density matrix, we could also increase the step size, which would reduce the number of iterations required to reach a certain
MPO multiplication approach (exponential)
If we wish to push the results to even larger
In other words, we can scan a range of exponentially increasing
βs_exp = 2.0 .^ (-3:3)
Z_analytic_exp = partition_function.(βs_exp, J, N)
F_analytic_exp = free_energy.(βs_exp, J, N)
Z_mpo_mul_exp = zeros(length(βs_exp))
# first iteration: start from high order Taylor expansion
ρ₀ = make_time_mpo(H, im * first(βs_exp) / 2, TaylorCluster(; N = 3))
Z_mpo_mul_exp[1] = double_logpartition(ρ₀)
# subsequent iterations: square
ρ = ρ₀
ρ_mps = convert(FiniteMPS, ρ₀)
for i in 2:length(βs_exp)
global ρ_mps, ρ
@info "Computing β = $(βs_exp[i])"
ρ_mps, = approximate(
ρ_mps, (ρ, ρ_mps), DMRG2(; trscheme = truncrank(D_max), maxiter = 10)
)
Z_mpo_mul_exp[i] = double_logpartition(ρ_mps)
ρ = convert(FiniteMPO, ρ_mps)
end
F_mpo_mul_exp = -(1 ./ βs_exp) .* Z_mpo_mul_exp
p_mpo_mul_exp_diff = let
labels = reshape(
map(expansion_orders[2:end]) do N
return "Taylor N=$N"
end, 1, :
)
p1 = plot(
βs, abs.(Z_taylor2 .- Z_analytic);
label = labels, title = "Partition function error", xlabel = "β", ylabel = "ΔZ(β)",
legend = :bottomright, yscale = :log10
)
plot!(p1, βs, abs.(Z_mpo_mul .- Z_analytic); label = "MPO multiplication")
plot!(p1, βs_exp, abs.(Z_mpo_mul_exp .- Z_analytic_exp); label = "MPO multiplication exp")
p2 = plot(
βs, abs.(F_taylor2 .- F_analytic); label = labels, xlabel = "β", ylabel = "ΔF(β)",
title = "Free energy error", legend = nothing, yscale = :log10
)
plot!(p2, βs, abs.(F_mpo_mul .- F_analytic); label = "MPO multiplication")
plot!(p2, βs_exp, abs.(F_mpo_mul_exp .- F_analytic_exp); label = "MPO multiplication exp")
plot(p1, p2)
end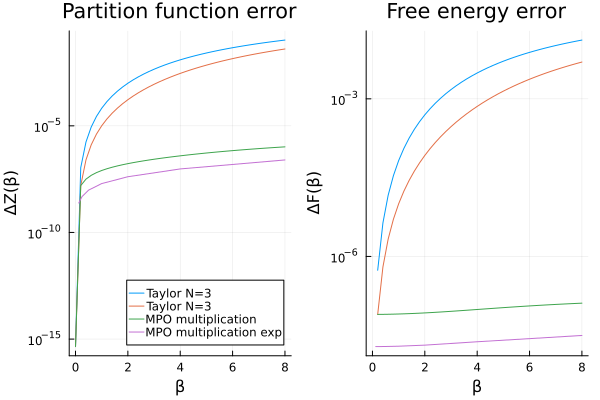
Clearly, the exponential approach allows us to reach larger
Warning
Again, using MPS techniques to approximate the multiplication of density matrices might lead to unphysical truncated density matrices. Increasing the stepsize could make this happen sooner, so we need to be careful with the maximal bond dimension.
Time evolution approach
Finally, we can also note that the partition function is characterized by the following differential equation:
In other words, we can compute the partition function at
The starting point for this approach could be either achieved through one of the techniques we have already discussed, but we can also start from the infinite temperature state directly. In particular, this state is given by the identity MPO, and we can evolve this state to compute the partition function at any
Z_tdvp = zeros(length(βs))
# first iteration: start from infinite temperature state
ρ₀ = infinite_temperature_density_matrix(H)
Z_tdvp[1] = double_logpartition(ρ₀)
# subsequent iterations: evolve by H
ρ_mps = convert(FiniteMPS, ρ₀)
for i in 2:length(βs)
global ρ_mps
@info "Computing β = $(βs[i])"
ρ_mps, = timestep(
ρ_mps, H, βs[i - 1] / 2, -im * (βs[i] - βs[i - 1]) / 2,
TDVP2(; trscheme = truncrank(64))
)
Z_tdvp[i] = double_logpartition(ρ_mps)
end
F_tdvp = -(1 ./ βs) .* Z_tdvp
p_mpo_mul_diff = let
labels = reshape(
map(expansion_orders) do N
return "Taylor N=$N"
end, 1, :
)
p1 = plot(
βs, abs.(Z_taylor2 .- Z_analytic); label = labels,
title = "Partition function error", xlabel = "β", ylabel = "ΔZ(β)",
legend = :bottomright, yscale = :log10
)
plot!(p1, βs, abs.(Z_mpo_mul .- Z_analytic); label = "MPO multiplication")
plot!(p1, βs_exp, abs.(Z_mpo_mul_exp .- Z_analytic_exp); label = "MPO multiplication exp")
plot!(p1, βs, abs.(Z_tdvp .- Z_analytic); label = "TDVP")
p2 = plot(
βs, abs.(F_taylor2 .- F_analytic); label = labels, xlabel = "β", ylabel = "ΔF(β)",
title = "Free energy error", legend = nothing, yscale = :log10
)
plot!(p2, βs, abs.(F_mpo_mul .- F_analytic); label = "MPO multiplication")
plot!(p2, βs_exp, abs.(F_mpo_mul_exp .- F_analytic_exp); label = "MPO multiplication exp")
plot!(p2, βs, abs.(F_tdvp .- F_analytic); label = "TDVP")
plot(p1, p2)
end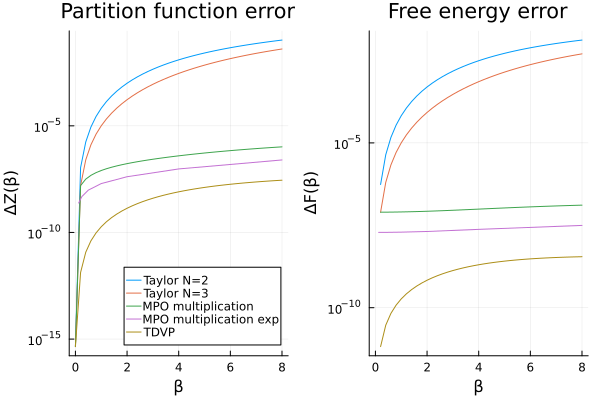
Note
We could further improve the accuracy of the TDVP approach by evolving with
This page was generated using Literate.jl.